


תזונה אסטרונאוטית: כיצד לשמור על בריאותך כמו אסטרונאוט בחלל
אסטרונאוטים נתקלים באתגרים רבים במהלך משימות חלל, ואחד האתגרים המשמעותיים ביותר הוא שמירה על תזונה מאוזנת ובריאה. התזונה בחלל דורשת תכנון מדויק, כאשר המזון צריך

מהו היהלום הטוב ביותר בחיתוך אמרלד?
כפי שציינו קודם, לקביעת חיתוך היהלומים הטוב ביותר עלינו לקחת בחשבון את ארבעת הקריטריונים שמציג ה-GIA להערכת היהלומים: דרגת חיתוך, משקל קראט, ניקיון וצבע. אין
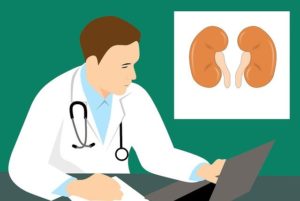
מהן אבני כליה? וכיצד הן משפיעות על בריחת שתן?
אבנים בכליות משפיעות על 1 מכל 11 אנשים בארה"ב. אבנים בכליות יכולות לכאוב מאוד. אבל מהן? מה גורם להן? ואיך אפשר למנוע אותן? המשיכו לקרוא

מדריך איך לבחור חברת גידור לחסכון בכסף
אז, אתה צריך להתקין גדר, אבל לא יודע איך לבחור חברת גדרות? האם אתה ממשיך לדחות את ההתקנה כי אתה דואג לגבי ההתחייבות הפיננסית? בחר

לימודים בקורס AutoCAD במכללת LFA
הצורך במומחים השולטים בתוכנת AutoCAD גדל מדי יום, ואת המחסור בהם רואים באתרי תעסוקה. ועכשיו הביקוש למומחי תכנון ופרויקטים יגדל עוד יותר, כי בישראל מתחילה
מהו הליך אישור אמ"ר?
כאשר יבואן מעוניין לייבא לישראל מוצרים מסוימים, אז אחד הדברים שהוא צריך לבדוק זה האם עליו לקבל אישורים מסוימים בשביל שהוא יוכל לייבא ולשווק אותם.

גודל סטנדרטי של שולחן אוכל
אם יש דבר אחד שכל מארח ומארחת מסכימים עליו, זה הכל על השולחן (בעקבות האוכל, כמובן). אחרי הכל, שולחן האוכל הוא המקום שבו המשפחה שלך

טיפים להתאמה אישית לארון לנוער
לתת לבני נוער מקום בארון שהם מרוצים ממנו בהחלט יכול להיות אתגר, אבל זה אתגר ששווה לקחת על עצמנו. ללא פתרונות אחסון פונקציונליים שהמתבגר שלך

כיצד לקפל כביסה כדי לחסוך מקום בארון ובמגירות?
אתה פשוט עורם את הבגדים וזורק אותם למגירה? האם פשוט נראה לך יותר נוח לתלות בגדים מאשר לקפל אותם? האם כל זה מתסכל אותך כי